“What is Marfan Syndrome?” The fibers that anchor and support the organs and other elements within your system are hereditary conditions that affect connective tissue. The center, uterus, blood vessels, and skeleton are often affected by Marfan syndrome.
In easy words, Marfan syndrome is a genetic disorder that affects the connective tissues in the body. Connective tissues provide support for the skin, bones, and organs. Marfan syndrome can cause a variety of problems, including heart defects, lung problems, and skeletal deformities.
With remarkably long arms, legs, fingers, and feet, people with Marfan syndrome are usually big and small. The injury could be moderate or severe because of Marfan syndrome. The disease may get life-threatening if your aorta, the large blood vessel that brings blood from the heart to the rest of your body, is damaged.
Take a look at the list of Famous People With Marfan Syndrome
Treatment generally requires drugs to maintain your blood pressure to reduce your aorta’s stress. It’s necessary to monitor to check for harm improvement occasionally. To fix the aorta, many individuals with this disease ultimately require preventive surgery.
Marfan Syndrome Symptoms

Even among members of the same household, its symptoms differ considerably. Some people only experience minor symptoms, but some develop life-threatening complications. In several other cases, with age, the disease continues to worsen.
Eye Complications

Over half of those with Marfan syndrome have eye difficulties, including nearsightedness (blurring of items much away), subluxation of the lens (the eye lens changes away from its usual position), or changes in eye shape or other eye issues.
Physical Look
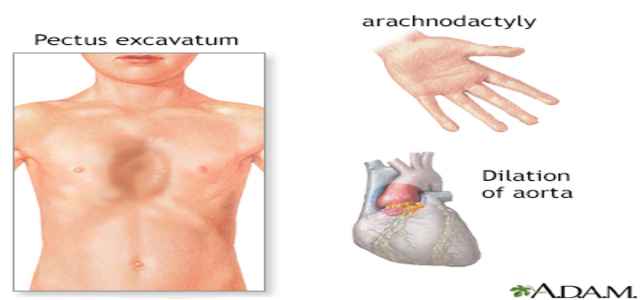
Additionally, there are quite tall and lean people with Marfan syndrome. They could look out of proportion with their arms, legs, fingers, and feet, also long for the remainder of their body. Their backbone could be flexed and stick out or be indented with their breastbone (sternum). Their joints may be brittle and displace easily.

There’s always a long, narrow face in people with this disease, and also, the roof of their mouth could be more significant than average, causing the teeth to the audience. Marfan syndrome causes many adjustments, especially dental and skeletal (bone) problems, to occur within the human body’s structures.
Cosmetic and Bone Infection
Patients with Marfan syndrome can have a dental background of thin palate tooth extractions or Illness expansions. Patients may also have a record of bone disorders, such as flat feet, hernias, and bone dislocations.
Alterations from the uterus, heart and blood vessels, skin, and lungs are different in this disease due to the twisted connective tissues.
Blood Vessel Changes
These blood vessels’ walls become shaky and dilate (extend ) with this disease. The aorta, the major artery that supplies blood to the heart to the rest of the human body, can also be influenced by those blood vessel modifications. An improved probability of aortic aneurysm, aortic dissection, or rupture (bursting) is present whenever the aorta’s walls stretch or weaken. It may dilate or dissect all areas of the aorta.
These conditions may lead to a health crisis and are life-threatening in some cases. Patients associated with Berry aneurysms may also have a history of intracranial bleeding or brain aneurysms.
Heart Valve Infection
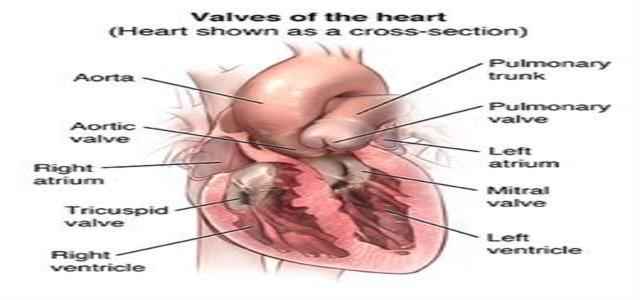
Marfan syndrome can help determine the heart’s valves, especially the mitral valve. The valve’s leaflets become floppy and don’t shut firmly, allowing blood to flow through the valve backward (mitral valve prolapse, also referred to as MVP). The valve leaks and the disease are known as mitral valve regurgitation because of MVP progress.
Additional Marfan Syndrome Signs
- A breastbone that protrudes external or slips inward
- A top, arched palate, and crowded teeth
- An abnormally curved spine
- Disproportionately long arms, legs, and fingers
- Intense nearsightedness
- Horizontal feet
- Heart murmurs
- Tall and slim build
Marfan Syndrome Causes
Reasons For Marfan syndrome encircle a gene defect that causes the entire body to make a protein, which tends to provide power and elasticity to connective tissues. Marfan syndrome is caused by a mutation in the FBN1 gene. This gene provides instructions for making a protein called fibrillin-1. This protein is found in connective tissues throughout the body. The FBN1 gene mutation alters the structure of fibrillin-1 proteins, which disrupts the organization of connective tissues. Marfan syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene is sufficient to cause the condition.
A parent with the illness inherits the abnormal gene from many people with Marfan syndrome. There’s a 50-50 chance for each child of an infected parent to inherit the faulty gene. The defective gene doesn’t come from either parent. About 25% of men and women have this disease. In such conditions, a new mutation develops.
Risk Factors & Complications
Marfan syndrome, which occurs in all races and cultural groups, affects people equally. The largest risk factor for Marfan syndrome is with a parent with the disorder, as it’s a hereditary condition.
Considering that virtually every portion of your body may be affected by Marfan syndrome, it may cause a broad selection of complications.
Skeletal Complications
Marfan syndrome increases the danger, such as scoliosis, of irregular spinal column curves. The organic rise of the ribs may also interfere, and this may activate the breastbone to protrude or seem to be sunken into your torso. With this disease, foot pain and low back pain are widespread.
Pregnancy Complications
The walls of the aorta, the main artery leaving the heart, might be ruined by Marfan syndrome. A lady’s heart pumps more blood than average during pregnancy, and this could put added stress on a woman’s aorta, which increases the probability of a fatal dissection or rupture.
Eye Complications
There are three Marfan syndrome complications when it comes to eye issues:
Lens Dislocation
If its strengthening structures weaken, then the lens within your eye will change weirdly. Ectopia lentis is the clinical term for this illness, and it occurs in over half of people who have this disease.
Retinal Problems
The danger of a tear or fracture in the retina, the light-sensitive tissue covering the rear of the eye, can also be improved by Marfan syndrome.
Early-Onset Glaucoma or Cataracts
These eye problems seem to happen at a younger age in people who have this disease. Glaucoma creates an increase in the eye’s pressure that could affect the optic nerve. From the eye’s generally clear lens, glaucoma is a muddy area.
Cardiovascular Infection
Aortic Aneurysm
Like a weak point in a tire, the blood pressure departing your heart can trigger the walls of your aorta to bulge out. It’s most likely to happen in the root cause, in which the artery exits the center, in people with this syndrome.
Aortic Dissection
Layers compose the walls of the aorta. Dissection occurs every time a minor tear in the surface of the aorta wall allows blood to squeeze between the wall’s outer and inner surfaces. This may result in large chest or back pain. An aortic dissection interrupts this boat’s look and may result in a rupture, which is catastrophic.
Valve Malformations
People who have this disease in their heart valves may have milder than normal tissue. This may generate valve tissue extending and irregular valve action. Your heart also has to work harder to compensate if heart valves don’t work appropriately. Afterward, this will end in heart failure.
Marfan Syndrome Diagnosis
As many connective tissue conditions have similar symptoms, Marfan syndrome may be stiff for physicians to diagnose. Additionally, the symptoms and signs of this syndrome change considerably among identical household members, both in their features and their seriousness.
To verify a diagnosis of Marfan syndrome, specific combinations of symptoms and family history have to be accessible. Someone might have any characteristics of this illness in some cases, although not enough of these to be diagnosed with this disease.
Marfan Syndrome Therapy

This syndrome necessitates a maintenance program tailored to the specifications of the individual. There could not be a medication required for specific men, just daily follow-up visits with their physician. Others might require surgery or drugs. The technique depends on the affected systems and the disease’s high level.
Do check out the list of Healthy Food Facts
Drugs
Medicines for the treatment of Marfan syndrome aren’t used but may be employed to prevent or handle complications. Medications could include:
A beta-blocker raises the heart’s ability to relax and reduces the pulse’s frequency and the pressure within the arteries, thereby quitting or diminishing the aorta’s expansion. Treatment with beta-blockers can begin from a young age.
A calcium channel blocker, such as verapamil, is proposed for reluctant people to take beta-blockers due to asthma or adverse outcomes.
A kind of drug acting on a chemical pathway in your system is an angiotensin receptor blocker (ARB). For the treatment of elevated blood pressure in addition to heart failure, these agents are also used. Clinical trials are now being conducted to ascertain how aortic expansion could be averted with these drugs.
Surgery
Marfan syndrome operation is targeted at preventing aortic dissection or rupture and fixing issues with the valve. Surgery is recommended if the aorta’s width is higher than 4.7 cm to 5.0 cm (dependent on the elevation ) or when the aorta is quickly expanding. Your cardiologist may also decide your aortic root diameter to height ratio because this might also influence whether you can have surgery. If you’re experiencing a pregnancy, it will also affect the operation suggestions.
Surgery guidelines are concentrated on aorta dimensions, called regular aorta dimensions, aortic growth rate, age, height, sex, and family history of aortic dissection. Surgery requires the replacement of graft for the dilated region of the aorta.
When Marfan syndrome causes, a leaky aortic or mitral valve (regurgitation) that tends to fluctuations in the left ventricle (lower left area of the heart) or heart failure, valve replacement or repair surgery might be required.
Final Words
Marfan syndrome is a disorder that affects the connective tissue in the body. Connective tissue helps to support the joints, bones, and organs. It can cause problems in many different parts of the body, including the heart, blood vessels, eyes, lungs, and skeleton.
This syndrome is caused by a mutation in the gene that encodes for fibrillin-1, a protein that is found in connective tissue. The severity of it varies from person to person. It is usually diagnosed in childhood or adolescence, but it can affect people of any age.
There is no cure for Marfan syndrome, but treatments are available to help manage the symptoms. With proper treatment, most people with this syndrome can lead normal, healthy lives.